News
Adding sequence-based identifiers to backbone taxonomy reveals 'dark taxa' fungi
Published 8/29/2018
Pilot project with northern European researchers enables inclusion of non-Linnaean ‘species hypotheses’ aimed at advancing scientific understanding of mycology and functional biodiversity

Until a few weeks ago, the GBIF backbone taxonomy fit snugly within a traditional model, classifying and ranking organisms' names using the system that Carl Linnaeus first outlined in Systema Naturae in 1735. By combining name-based information from dozens of different authoritative sources like the Catalogue of Life, IRMNG and the World Register of Marine Species (affectionately known as 'WoRMS'), the backbone provides a consistent means of organizing all species-related content on GBIF.org—like datasets, occurrences and species pages—and enables all forms of taxonomic searching, browsing and reporting.
But in recent months, hidden forces have been at work, extending the backbone's capacity to accommodate an emerging taxonomic paradigm. Working with partners in Estonia, Sweden, Denmark and other northern European countries, GBIF has successfully piloted the publication of 11,495 fungal occurrence records linked to DNA-based identifiers that appear in the latest release of the GBIF backbone taxonomy.
What's the (dark) matter with fungi?
For some taxonomic groups, advances in molecular sequencing have made it increasingly possible to observe and examine organisms and ecological communities using DNA-based sequences alone. Many organisms observed through DNA metabarcoding are new to science, and some represent new higher-order lineages up to the phylum level, and possibly beyond.
But without physical specimens or accepted scientific names, these sequences cannot not be linked to the Linnaean-based nomenclature Codes that set the rules governing the biological classification system. Lacking formally accepted taxonomic identities—or, more simply, scientific names—these 'dark' or 'dark matter' taxa are prone to being overlooked not just in scientific research, but even legal contexts.
The hyperdiverse kingdom of Fungi hosts untold quantities of cryptic biodiversity. Mycology researchers recognize that a huge gap exists between the numbers of described and actual species—estimates suggest, in fact, that many times more fungi species exist than have been described to date. However, the practice of assigning formal scientific names hinges on the thorough analysis and description of a given organism's form and structural features—a process that is harder to apply to fungal species than to plants and animals.
For example, the mushrooms that most equate with species of fungi are only fruiting bodies, organs of sexual reproduction. Given that our knowledge of fungal reproductive cycles and sexual stages is often extremely limited, expecting to find specimens of these for all species easily and anytime we like is no more reasonable than expecting to find apple trees that give fruit year-round. Sequencing provides a means of detecting the presence of fungal species in the absence of fruiting bodies.
Bringing order to dark taxa
In hope of addressing this gap in scientific knowledge, experts from the University of Tartu Natural History Museum (home to the GBIF Estonia node) and the University of Gothenburg developed UNITE, a system that uses ribosomal DNA-based sequences to give an identity to these cryptic elements of fungal biodiversity. The main building block for UNITE is the 'species hypothesis' (SH), which groups similar sequences into provisional species-level clusters.
Each species hypothesis is assigned a DOI, establishing a stable, permanent reference for that particular hypothesis (sequences known to derive from organisms already in possession of formally described Linnaean scientific names can, of course, rely on those). For sequences known only from environmental samples, this system has the advantage of giving unambiguous identities to species hypotheses.
Researchers can cite individual hypotheses, and those who discover the same hypotheses can relate them to each another, assembling in the process metadata that can facilitate and expedite the formal description of the underlying species. In this way, species hypotheses are meant not to replace the Linnaean enterprise but to increase the speed of species discovery and description.
The approach has led to the definition of more than 73,000 fungal species hypotheses, which together rely on and combine more than half of the system's 817,130 public reference DNA sequences.
By relying on traditional Linnaean tools like molecular keys, reference sequences, and taxonomically specified vouchers while accounting for taxonomic uncertainty, the species hypothesis provides a well-ordered operational taxonomic unit (OTU) within the framework of UNITE.
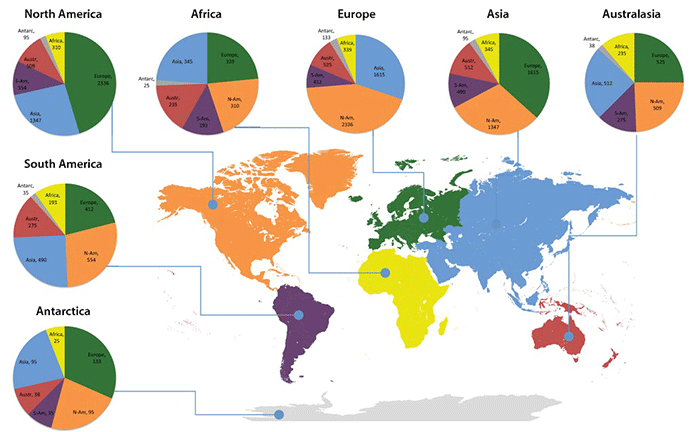
Circle graphs illustrate the geographical distribution of UNITE species hypotheses that occur on more than one continent. North America, Europe and Asia are more similar to each other compared with other continents. The comparatively high number of shared SHs between Southern and Northern Hemisphere continents mark potential invasions that suggest the need for fine-scale ecological studies. Detail from Figure 2, Kõljalg U et al. (2013).
Testing (species) hypotheses with sequence-based occurrences
With the growing use of UNITE by mycologists working with molecular data, the checklist containing all names in UNITE provides nearly all the necessary preconditions for testing whether and how sequence-based identifications could appear alongside traditional Linnaean names. All that was missing was a set of occurrences to match to the checklist’s catalogue of formally described species and proposed species hypotheses.
Enter the BIOWIDE, a nation-wide collaboration between Aarhus University, the University of Copenhagen, the Natural History Museum of Aarhus, and the National History Museum of Denmark. Started in 2014, the project strived to live up to its full name (BIOdiversity in WIdth and DEpth) through surveys that collected a comprehensive range of environmental data from 130 terrestrial sampling sites across Denmark. Included among these were bulk soil samples from each site sequenced for fungi and identified by UNITE species hypotheses.
On the face of it, there's little to differentiate the BIOWIDE eDNA Fungi dataset from other occurrence datasets. But start digging into the occurrences, and you'll find species hypothesis (SH) numbers noting affinities and relations with Linnaean species concepts littering the lists. In fact, just 769 of the dataset's 2,680 mycological identifiers correspond to formally described scientific names. The remaining 1,911 are all classified under a species hypothesis from UNITE.
Why dark taxa matter
While this experiment is enabled by data, the aim is squarely on advancing science. For starters, researchers may discover pervasive but previously unknown species. For example, the BIOWIDE team found in sequences of this undescribed species in the fungal order of Cantharellales ranging across a dozen disparate plots in Denmark. How far does the range of this undescribed species extend?
The test also highlights the potential for implications at the science-policy interface. Searching BIOWIDE for Hygrocybe conica, commonly known as witch's hat, yields eight occurrences across on two species hypotheses: SH176829.07FU and SH176834.07FU. The taxonomy of H. conica reveals dozens of synonyms associated with this name, and no fewer than nine species hypotheses, suggesting that the concept is actually a species aggregate. But when the Danish national Red List describes Kegle-vokshat as a species of “Least concern”, to which species—or species hypothesis—does this refer?
Surfacing taxa like these fungi is critically important to science as well, because these underrepresented groupings represent the 'dark matter' of biodiversity and have key functional roles and effects in ecological communities and ecosystems. By enabling scientists to explore microbiomes in which many or most organisms may never relate to an accepted species name, metagenomic sampling offers the possibility of detecting and providing molecular identification of taxa with unheard-of scale and speed—not just for fungi, but also soil- and ocean-sediment bacteria, plankton and larval invertebrates, among others.
By combining elements of morphological and molecular biodiversity in a single taxonomic backbone, using principles common to both the Linnaean and metagenomic paradigms, the GBIF network is better positioned to address major spatial, temporal and taxonomic gaps and bias while supporting scientific research to understand functional biodiversity.